GMP aktuell
Was bedeutet GMP?
Warum ist GMP wichtig?
Wann wurde GMP eingeführt?
Die ersten Regeln zur Sicherstellung der Arzneimittelqualität wurden in den USA bereits Anfang des 20. Jahrhunderts aufgrund von Problemen mit der Arzneimittelqualität eingeführt. Immer neue Problemfälle bzw. nach heutiger Bezeichnung Skandale führten zu immer stärker regulierenden Vorschriften für die Hersteller von Arzneimitteln. In Deutschland wurde 1975 das Bundesgesundheitsamt (BGA) gegründet, das 1994 in das Bundesinstitut für Arzneimittel und Medizinprodukte (BfArM) umgewandelt wurde. Seit 1976 gibt es das Arzneimittelgesetz (AMG), von 1985 bis 2006 zusätzlich die Pharmabetriebsverordnung (PharmBetrV), seit 2006 die Arzneimittel und Wirkstoff Herstellungsverordnung (AMWHV).
Wer erlässt GMP Richtlinien?
Grundsätzlich kann jedes Land seine eigenen Regeln festlegen. Als Folge der Internationalisierung der Herstellung von Arzneimitteln (Stichwort Lieferketten) und des Handels mit Arzneimitteln gibt es inzwischen gemeinsame, harmonisierte Standards, z.B. von der WHO. In der Europäischen Union sind die GMP Richtlinien in Form von Richtlinien (Directives, müssen von den Mitgliedsstaaten in nationales Recht „übersetzt“ werden), Regulations (Verordnungen, gelten unmittelbar 1:1 in den Mitgliedstaaten) und Leitlinien (Guidelines) festgelegt.
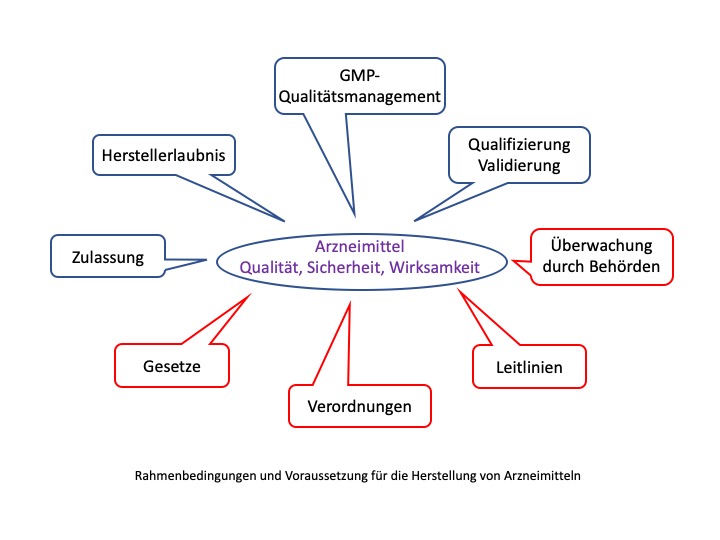
Wo finden sich GMP Anforderungen?
In Deutschland sind die GMP Anforderungen in Form von Gesetzen (z.B. Arzneimittelgesetz) und Verordnungen (z.B. Arzneimittel und Wirkstoff Herstellungsverordnung) verankert. Zusätzlich gibt es Leitlinien, die nicht in Form von Gesetzen oder Verordnungen verankert sind.
Sind GMP Leitlinien verbindlich?
Gibt es eine GMP Zertifizierung?
Arzneimittelhersteller und -prüflaboratorien unterliegen der Überwachung durch Aufsichtsbehörden. In Deutschland erfolgt diese Überwachung dezentral z.B. durch die Regierungspräsidien in den Bundesländern. In den USA erfolgt die Überwachung durch die FDA (Food and Drug Administration). Sie überwacht auch Hersteller in anderen Ländern, die ihre Arzneimittel auf den US-Markt bringen wollen. Ähnliche Behörden gibt es auch in anderen Ländern. Diese Behörden erteilen dann Erlaubnisse und Zertifikate, die zur Herstellung bzw. Prüfung von Arzneimitteln berechtigen. In Deutschland braucht jeder Hersteller von Arzneimitteln eine Herstellerlaubnis, in der auch die Arzneimittel aufgelistet sind, die hergestellt werden dürfen. Diese Erlaubnisse und Zertifikate haben eine begrenzte Gültigkeit. Die Überwachungsbehörden führen regelmäßige Inspektionen durch, in denen überprüft wird, ob der jeweilige Hersteller oder das Prüflabor die Anforderungen weiterhin erfüllt.
Zusätzlich muss für jedes Arzneimittel eine produktspezifische Zulassung beantragt werden. Dies geschieht für Europa meist bei der Europäische Arzneimittel Agentur (EMA). Dazu müssen umfangreiche Daten zum Nachweis von Qualität, Wirksamkeit und Sicherheit des Arzneimittels eingereicht werden. Die EMA kann dann die Zulassung empfehlen, die durch die Europäische Kommission beschlossen und erteilt wird. Nationale Zulassungen erteilt in Deutschland das BfArM. Nur wenn eine Zulassung erteilt wurde, darf ein Arzneimittel auf den Markt gebracht werden. Für bestimmte Arzneimittel, z.B. Impfstoffe, muss außerdem noch jede einzelne Charge des Arzneimittels durch eine Behörde geprüft und freigegeben werden. In Deutschland ist hierfür das Paul-Ehrlich-Institut (PEI) zuständig
Wo finde ich praktische GMP Standards?
Die Gesetze, Verordnungen und Leitlinien beschreiben, was die Anforderungen und Standards sind, also was ein Arzneimittelhersteller oder -prüflabor sicherstellen muss. Die Anforderungen sind so geschrieben, dass sie für eine Vielzahl an unterschiedlichen Unternehmen anwendbar sein sollen. Daher lassen sie an vielen Stellen Interpretationsspielraum, wie die Anforderungen praktisch erfüllt werden können.
Hilfreiche Hinweise zur praktischen Umsetzung finden sich in Leitlinien verschiedener Organisationen, auf entsprechenden Internetplattformen und in Büchern, wie z.B. dem GMP-Berater aus dem GMP-Verlag.
Was ist GMP-Qualitätsmanagement?
Was ist GMP-Qualitätsmanagement?
Das GMP-Qualitätsmanagement einhaltet alle Prozesse und Systeme die zur Qualität, Sicherheit und Wirksamkeit eines Arzneimittels beitragen. Dazu gehören z.B. aber nicht ausschließlich
- Dokumente, die Vorgaben für Prozesse beschreiben
- Dokumente, die die korrekte Ausführung der Prozesse nachweisen
- Die Kontrolle von Änderungen an Prozessen
- Die Bewertung von Abweichungen von den Vorgaben, von Fehlern und von Reklamationen
- Maßnahmen zur Verbesserung von Prozessen und Systemen
Den Bereichen Herstellung (inklusive Lagerung und Distribution), Qualitätskontrolle und Qualitätssicherung kommen dabei wichtige Rollen zu.
Was ist GMP-Risikomanagement?
Ein wichtiger Bestandteil des GMP Qualitätsmanagements ist das Risikomanagement mit den Risikoanalysen. Sie sollen bewirken, dass die Arzneimittelhersteller mögliche Risiken für den Patienten bzw. Anwender identifizieren, bewerten und durch geeignete Maßnahmen minimieren Dabei werden zunächst mögliche Risiken identifiziert und dann deren Auftrittswahrscheinlichkeit verringert und die Entdeckungswahrscheinlichkeit erhöht.
Zur Risikoanalyse gibt es verschiedene Methoden, die je nach Anwendungsbereich und Komplexität des betrachteten Themas eingesetzt werden können.
Was sind GMP-Qualifizierung und -Validierung?
Will ein Arzneimittelhersteller ein Gebäude, eine Anlage oder einzelne Geräte nutzen, so muss er sicherstellen und nachweisen, dass die Gebäude, Anlagen und Geräte für den gewünschten Zweck geeignet sind, also die allgemeinen und produktionsspezifischen Anforderungen erfüllen. Das gleiche gilt für Prüflaboratorien. Dieser Nachweis geschieht im Rahmen der Qualifizierung. Es kann also nicht einfach ein entsprechendes Gerät gekauft oder konstruiert werden und dann sofort eingesetzt werden. Je nach Komplexität des Gerätes oder der Anlage kann die Qualifizierung einen erheblichen Umfang an Ressourcen, Dokumentation und Zeit erfordern.
Die Prozesse, die dann auf den qualifizierten Anlagen und Geräten laufen, müssen validiert werden. Validierung bedeutet, dass ich nachweise, dass mein Prozess innerhalb der vorgesehenen Verfahrensgrenzen reproduzierbar die erforderliche Qualität des Arzneimittels liefert. Auch dies erfordert einen erheblichen Umfang an Ressourcen, Dokumentation und Zeit.
Was sind GMP Reinraumklassen?
Die GMP Reinraumklassen definieren Anforderungen an die Reinheit von Räumen hinsichtlich der Luftqualität und der mikrobiologischen Belastung von Oberflächen. Gerade für die Herstellung von sterilen Arzneimitteln, die injiziert werden müssen (z.B. Impfstoffe) ist dies von enormer Bedeutung.
Das fängt bei der Konstruktion der Reinräume an (glatte Oberflächen ohne unnötige Ecken, Kanten, Winkel, gute Reinigbarkeit und gute Desinfektionsmittelbeständigkeit der Oberflächen), erfordert leistungsfähige Belüftungssysteme für die Reinräume, die auch kleinste Partikel und Mikroorganismen aus der Luft filtern, dies betrifft ebenso die Konstruktion der Geräte, die gut zu reinigen und zu desinfizieren sein müssen und im Betrieb nicht unnötig Partikel absondern dürfen. Die Arbeit in Reinräumen erfordert außerdem speziell ausgebildetes Personal, das entsprechende Bekleidungs- und Hygienevorschriften einhält.
